ОРИГИНАЛЬНОЕ ИССЛЕДОВАНИЕ
Выделение и характеристика бактериофагов Pseudomonas aeruginosa — потенциальных агентов для фаговой терапии
Федеральный научно-клинический центр физико-химической медицины Федерального медико-биологического агентства, Москва, Россия
Одним из патогенов, характеризующихся критическим показателем доли штаммов с множественной лекарственной устойчивостью (МЛУ), является Pseudomonas aeruginosa. В качестве альтернативы антибиотикам при терапии инфекций, вызванных штаммами с МЛУ, рассматривают фаготерапию. Целью исследования было выделить и охарактеризовать бактериофаг P. aeruginosa, потенциально пригодный для терапии инфекционных заболеваний. Выделение проводили методом накопительных культур. Спектр литической активности устанавливали спот-тестированием на коллекции из 40 штаммов P. aeruginosa. Полногеномное секвенирование выполняли на платформе MiSeq (Illumina). Филогенетический анализ геномов проводили с помощью VICTOR. Выделенные бактериофаги vB_PaeA-55-1w и vB_PaeM-198, принадлежащие к семействам Autographiviridae и Myoviridae соответственно, обладали широким спектром литической активности (около 50% каждый), в том числе вызывали лизис штаммов с МЛУ. Геномы vB_PaeA-55-1w и vB_PaeM-198 представлены двухцепочечной ДНК длиной 42,5 и 66,3 т.п.н. соответственно. В составе геномов аннотировано 52 (vB_PaeA-55-1w) и 95 (vB_PaeM-198) открытых рамок считывания, среди них гены интеграз и токсинов не обнаружены. На филогенетическом древе vB_PaeA-55-1w располагался в кластере совместно с бактериофагами рода Phikmvvirus семейства Autographiviridae, в том числе с используемыми в фаготерапии, а vB_ PaeM-198 входил в кластер, включающий бактериофаги рода Pbunavirus семейства Myoviridae. Бактериофаги vB_PaeA-55-1w и vB_PaeM-198 можно рассматривать в качестве кандидатов для применения в фаготерапии, в том числе и для лечения инфекций, вызванных штаммами P. aeruginosa с МЛУ.
Ключевые слова: полногеномное секвенирование, филогенетический анализ, Pseudomonas aeruginosa, вирулентные бактериофаги, фаготерапия, Autographiviridae, Myoviridae
Финансирование: исследование выполнено за счет средств, предоставленных для выполнения государственного задания «Разработка персонализированного подхода терапии инфекционных процессов с применением вирулентных бактериофагов» (ШИФР: Бактериофаг).
Благодарности: авторы благодарят Центр высокоточного редактирования и генетических технологий для биомедицины ФГБУ ФНКЦ ФХМ ФМБА России за секвенирование геномов бактериофагов.
Вклад авторов: М. А. Корниенко — план исследований, набор и обработка данных, написание статьи; Н. С. Купцов — набор и обработка данных, написание статьи; Д. И. Данилов, Р. Б. Городничев, М. В. Малахова, В. А. Веселовский — набор данных; Д. А. Беспятых — обработка данных, Е. А. Шитиков — план исследований, обработка данных, написание статьи; Е. Н. Ильина — план исследований, написание статьи.
Соблюдение этических стандартов: экспериментальная работа выполнена с соблюдением норм Санитарно-эпидемиологических правил «Безопасность работы с микроорганизмами III–IV групп патогенности (опасности) и возбудителями паразитарных болезней» СП 1.3.2322-08; Санитарно-эпидемиологических правил СП 1.3.2518-09 — «Дополнения и изменения № 1 к санитарно-эпидемиологическим правилам «Безопасность работы с микроорганизмами III–IV групп патогенности (опасности) и возбудителями паразитарных болезней» СП 1.3.2322-08; Санитарноэпидемиологических правил «Санитарно-эпидемиологические требования к обращению с медицинскими отходами» СанПиН 2.1.7.2790-10, а также Федеральных клинических рекомендаций «Рациональное применение бактериофагов в лечебной и противоэпидемической практике».
Для корреспонденции: Мария Андреевна Корниенко
ул. Малая Пироговская, д. 1а, г. Москва, 119435; moc.liamg@ayiramokneinrok
По данным Всемирной организации здравоохранения, смертность от устойчивых к антибиотикам бактерий находится на историческом максимуме [1]. При этом первые места в списке патогенов, представляющих наибольшую угрозу для здоровья человека, занимают грамотрицательные бактерии, в том числе Pseudomonas aeruginosa [1]. Бактерии этого вида распространены повсеместно, обладают высокой степенью генетической пластичности и приспособляемости к условиям окружающей среды. Инвазия штаммами P. aeruginosa нередко приводит к генерализации инфекционного процесса за счет разнообразия механизмов патогенности [2]. P. aeruginosa является причиной широкого спектра заболеваний — от интоксикации до обширных гнойновоспалительных процессов и септического шока [2]. Стабильно занимая лидирующие позиции среди возбудителей нозокомиальных инфекций в России, доля изолятов P. aeruginosa среди всех бактериальных возбудителей внутрибольничных инфекций, выделенных за 2015–2020 гг., по данным портала AMRmap, составила, 16,83% [3]. Кроме того, для P. aeruginosa характерна высокая доля штаммов в популяции, обладающих множественной лекарственной устойчивостью (МЛУ) (штаммы, устойчивые по крайней мере к одному антибиотику из трех и более групп антибиотиков) (около 30%), а также экстремальной лекарственной устойчивостью (ЭЛУ) (штаммы, устойчивые по крайней мере к одному антибиотику из всех групп антибиотиков, за исключением 1–2 групп) (около 15%), что является одним из факторов высокой смертности пациентов [4].
На сегодняшний день первостепенной задачей становится разработка альтернативных антибиотикам методов терапии инфекционных заболеваний, вызванных патогенами с МЛУ и ЭЛУ. Применение препаратов вирулентных бактериофагов для терапии инфекционных процессов (фаготерапия) является одним из наиболее перспективных вариантов [5]. Это обусловлено тем фактом, что вирулентные бактериофаги лизируют как чувствительные, так и устойчивые к антибиотикам штаммы бактерий. Кроме того, применение бактериофагов не вызывает побочных токсических и аллергических реакций и не имеет противопоказаний [6], а также показано для терапии беременных женщин в сочетании с другими лечебными препаратами [7].
Успех фаговой терапии для лечения инфекций P. aeruginosa у животных и людей был подтвержден в ряде публикаций, а также в доклинических и клинических испытаниях [8, 9]. В Российской Федерации выпускают ряд коммерческих терапевтических препаратов для лечения инфекций, вызванных P. aeruginosa: «Бактериофаг синегнойный», «Интести-бактериофаг», «Пиобактериофаг поливалентный очищенный» («Микроген»; Россия).
Несмотря на наличие препаратов бактериофагов, активных по отношению к P. aeruginosa, и успешный опыт их применения, необходима постоянная работа по обновлению фаговых коллекций, что связано со спецификой современной фаготерапии. Так как вирулентные бактериофаги имеют довольно узкую специфичность, обычно направленную на ряд штаммов, обновление коллекции позволит включить туда фаги, вызывающие лизис актуальных бактериальных штаммов. Кроме того, описан ряд случаев возникновения устойчивых к бактериофагам мутантных форм бактерий [10]. Выделение новых бактериофагов и включение их в состав терапевтических препаратов решают данную проблему.
В связи с вышеизложенным цель данной работы — выделение и характеристика бактериофагов P. aeruginosa, потенциально пригодных для терапии инфекционных заболеваний.
МАТЕРИАЛЫ И МЕТОДЫ
Бактериальные штаммы
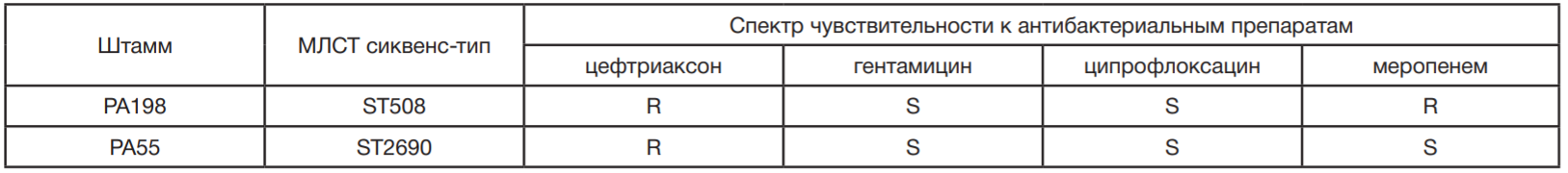
Исследование проводили на штаммах P. aeruginosa (n = 40), отобранных из коллекции бактериальных штаммов лаборатории молекулярной генетики микроорганизмов Федерального научно-клинического центра физикохимической медицины Федерального медико- биологического агентства России. Штаммы коллекции были охарактеризованы по профилю лекарственной чувствительности (к цефтриаксону, гентамицину, ципрофлоксацину и меропенему), а также генотипам согласно результатам мультилокусного секвенирования — типирования (МЛСТ) [11]. Культивирование бактерий проводили в течение 18–24 ч на питательной среде LB (от англ. lysogeny broth) (Oxoid; Великобритания) при температуре 37 °С.
Выделение бактериофагов
Бактериофаги выделяли методом накопительных культур из природных источников (сточные вод, проб воды из различных рек) на штаммах P. aeruginosa PA55 и PA198 (табл. 1). Для этого пробу воды в объеме 50 мл пропускали через фильтр Millipore с мембраной из поливинилиденфторида и диаметром пор 0,45 мкм (Merck Millipore; США) и добавляли к ней двукратный бульон LB. В дальнейшем вносили 300 мкл ночной культуры штаммахозяина и инкубировали на качалке при 37 °С в течение 18 ч. После культивирования бактериальные клетки осаждали центрифугированием при 3500 g в течение 10 мин, надосадок пропускали через фильтр Millipore с мембраной из полиэфирсульфона и диаметром пор 0,22 мкм (Merck Millipore; США). Индивидуальные бактериофаги получали последовательным (трехкратным) выделением из отдельных негативных колоний. В дальнейшем бактериофаги наращивали в 50 мл бульона LB, содержащего 300 мкл ночной культуры бактериального штамма. Концентрацию бактериофага в фаговом лизате оценивали стандартным методом титрования по Грациа [12].
Определение спектра литической активности бактериофагов
Определение спектра литической активности бактериофагов осуществляли методом спот-тестирования (от англ. spot). С целью предотвращения неспецифического лизиса для тестирования использовали фаголизаты с титром 3 × 106 БОЕ/мл (бляшкообразующих единиц в мл). Ночную культуру (1010 КОЕ/мл (колониеобразующих единиц в мл)) тестируемого бактериального штамма последовательно разводили в бульоне LB до концентрации клеток 106 КОЕ/мл, смешивали 0,1 мл с 5 мл полужидкого LB-агара (0,6% агара) и вносили в чашку Петри, содержащую тонкий слой LB агара (1,5%-го агара). После застывания полужидкого агара на поверхность чашки наносили каплю (5 мкл) исследуемого бактериофага. Чашки Петри инкубировали при температуре 37 °С в течение 18–24 ч. Литическую активность оценивали визуально: бактериальный штамм считали чувствительным к действию бактериофага в случае образования прозрачного пятна либо отдельных негативных колоний. При отсутствии пятна лизиса либо наличии мутного пятна бактериальный штамм относили к устойчивым штаммам.
Выделение ДНК бактериофагов и полногеномное секвенирование
Тотальную ДНК бактериофагов выделяли методом фенолхлороформной экстракции [13] с предварительной ферментативной обработкой фаговых лизатов РНКазой А, ДНКазой I и протеиназой К (Thermo Fisher Scientific; США) в соответствии с инструкцией производителя.
Для приготовления библиотеки было взято 250 нг геномной ДНК. Фрагментирование ДНК проводили на приборе Covaris S220 System (Covaris; США) до размера (400–500 п.н.). Качество фрагментированных образцов оценивали на биоанализаторе Agilent 2100 (Agilent; США) в соответствии с инструкцией производителя. Набор NEBNext Ultra II DNA Library Prep Kit (New England Biolabs; США) использовали для подготовки геномных библиотек, а набор NEBNext Multiplex Oligos kit for Illumina (96 индексных праймеров, New England Biolabs; США) — для индексации библиотек. Количественный анализ библиотек проводили с помощью набора Quant-iT DNA Assay Kit, High Sensitivity (Thermo Scientific; США). Процедуру секвенирования осуществляли с помощью инструмента MiSeq с использованием набора реагентов для секвенирования MiSeq Reagent Nano Kit v2 (500cycle) (Illumina; США) согласно рекомендациям производителя.
Биоинформатический анализ геномов бактериофагов
Сборку полногеномных последовательностей бактериофагов выполняли с использованием программного пакета Prokka v1.14.6 [14]. Аннотации геномов бактериофагов получали с помощью Rapid Annotation Using Subsystem Technology (RAST) [15]. Функции некоторых открытых рамок считывания (ОРС) предсказывали с помощью
BLASTP (https://blast.ncbi.nlm.nih.gov/Blast.cgi) и HHpred (https://toolkit.tuebingen.mpg.de/#/tools/hhpred). Поиск транспортных РНК (тРНК) осуществляли с помощью программы ARAGORN [16]. Полученные геномы были депонированы в базу данных GenBank под номерами MZ553931 и MZ553930 для бактериофагов vB_PaeA-551w и vB_PaeM-198 соответственно.
Таксономическую принадлежность исследуемых бактериофагов определяли на основании гомологии их геномных последовательностей с последовательностями бактериофагов, представленных в базе данных GenBank с помощью сервиса BLASTN (https://blast.ncbi.nlm.nih.gov/ Blast.cgi). В зависимости от таксономического положения бактериофагам присваивали названия в соответствии с рекомендациями ICTV (от англ. International Committee on Taxonomy of Viruses) [17].
Филогенетический анализ геномов проводили с помощью онлайн-инструмента VICTOR, используя метод GBDP (Genome-BLAST Distance Phylogeny) с рекомендованными настройками для вирусов прокариотов [18]. Обработку ветвей проводили с помощью FASTME [19] по формуле D0, визуализацию — с помощью FigTree [20]. В случае бактериофага vB_PaeA-55-1w в сравнение включали геномы бактериофагов со следующими номерами в базе данных GenBank: NC_054890, NC_047953, NC_047967, NC_048201, NC_047965, NC_026602, NC_027375, NC_047956, NC_047957, NC_031014, NC_016764, NC_030923, NC_028836, NC_013638, NC_004665, NC_047922, NC_015264, NC_012418, NC_047827, NC_009936, NC_009935, NC_010326, NC_022746, NC_022091, NC_047955, NC_047852, NC_047954, NC_047933, NC_047952, NC_027292, MG250485, NC_047997, NC_015208, NC_005045, NC_047747, NC_024362, NC_021062, NC_042104, NC_028661, NC_023005, NC_047981, NC_011107, NC_011105, NC_048168, NC_048200, NC_047894, NC_047873, NC_041885, NC_047826. Для бактериофага vB_PaeM-198 использовали следующие геномы: NC_048675, NC_048744, NC_048626, NC_048662, NC_048663, NC_048676, NC_048745, NC_011703, NC_026587, NC_026600, NC_042113, NC_019918, NC_028745, NC_028971, NC_048109, NC_041870.1, NC_042080, NC_042079, NC_007623, NC_007810, NC_041968, NC_019450, NC_017674, NC_042054, NC_030934, NC_015272, NC_019935, NC_041865, NC_011165, NC_011166, NC_048699.1, NC_031073, NC_031073, MN871467, NC_041994, NC_041903, NC_041902.1, NC_042060, NC_048806, NC_022096, NC_015294, NC_022967.1, NC_022966, NC_022986, NC_019913, NC_011810, NC_041881, NC_030940, NC_003278, NC_004629, NC_031110, NC_041880, NC_028882, NC_028939, NC_023601, NC_042081, NC_042081, NC_042081, NC_011756, NC_042092, NC_029065, NC_041904.
Модульное строение геномов было определено на основании аннотации, а также в ходе определения гомологии нуклеотидных последовательностей отдельных ОРС с помощью BLASTN (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Выделение бактериофагов и характеристика спектра литической активности
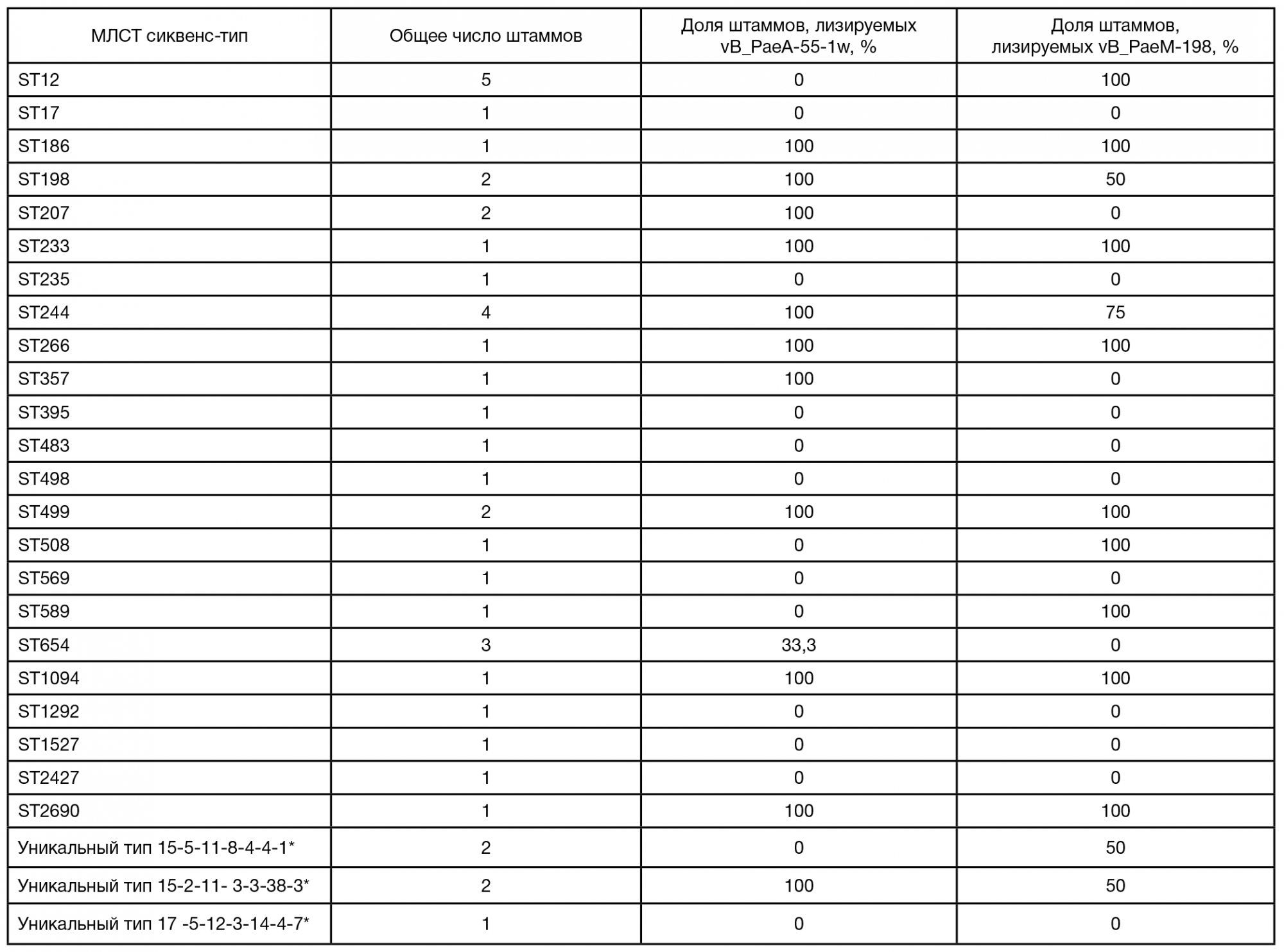
Для выделения бактериофагов, активных в отношении бактерий вида P. aeruginosa, из коллекции бактериальных штаммов ФГБУ ФНКЦ ФХМ ФМБА России были выбраны штаммы PA55 и PA198 (см. табл. 1). На данных штаммаххозяевах были получены два бактериофага, впоследствии названные vB_PaeA-55-1w и vB_PaeM-198. Согласно исследованию спектров литической активности, фаг vB_PaeA-55-1w вызывал лизис 19 штаммов коллекции (47,5%), в то время как vB_PaeM-198 — 20 штаммов (50%) (табл. 2). Для выделения бактериофагов, активных в отношении бактерий вида P. aeruginosa, из коллекции бактериальных штаммов ФГБУ ФНКЦ ФХМ ФМБА России были выбраны штаммы PA55 и PA198 (см. табл. 1). На данных штаммаххозяевах были получены два бактериофага, впоследствии названные vB_PaeA-55-1w и vB_PaeM-198. Согласно исследованию спектров литической активности, фаг vB_PaeA-55-1w вызывал лизис 19 штаммов коллекции (47,5%), в то время как vB_PaeM-198 — 20 штаммов (50%) (табл. 2). Следует также отметить, что из 17 штаммов коллекции, обладающих МЛУ, бактериофаг vB_PaeA-55-1w лизировал 8 штаммов (47%), а бактериофаг vB_PaeM198 — 6 штаммов (35%).
Полногеномное секвенирование бактериофагов
Для детальной характеристики исследуемых бактериофагов были получены данные полногеномного секвенирования и проведена их аннотация. Геномы бактериофагов были представлены двухцепочечной ДНК и имели длину 42,5 т.п.н. (vB_PaeA-55-1w) и 66,3 т.п.н. (vB_PaeM-198). Бактериофаг vB_PaeA-55-1w кодировал 52 ОРС, в то время как vB_PaeM-198 — 95. Для анализируемых бактериофагов не показано наличие в геноме тРНК.
В ходе сопоставления полученных полногеномных последовательностей с геномами, доступными в базе данных Genbank, было установлено таксономическое положение бактериофагов, а также их ближайший родственник. Бактериофаг vB_PaeA-55-1w относился к роду Phikmvvirus семейства Autographiviridae, а наиболее близкий геном соответствовал фагу Pseudomonas phage MYY9 (процент идентичности — 95%, длина выравнивания — 97,59%, номер в базе Genbank — MW406975.1). Бактериофаг vB_PaeM-198 принадлежал к роду Pbunavirus семейства Myoviridae и имел высокий процент идентичности с фагом Pseudomonas phage phiKT28 (процент идентичности — 99%, длина выравнивания — 96,34%, номер в базе Genbank — KP340287.1).
Филогенетический анализ бактериофагов Pseudomonas spp
Для позиционирования исследуемых бактериофагов внутри соответствующих семейств использовали референсные геномы, рекомендованные ICTV [17]. В филогенетический анализ семейства Autographiviridae было включено 50 геномов бактериофагов, активных против Pseudomonas spp (рис. 1А), для семейства Myoviridae — 60 геномов (рис. 1Б).
На филогенетическом древе бактериофагов Pseudomonas spp. семейства Autographiviridae можно выделить два больших кластера (см. рис. 1А). Первый кластер включает бактериофаги, хозяевами которых являются бактерии видов P. aeruginosa, а также по одному бактериофагу Pseudomonas agarici, Pseudomonas putida, Pseudomonas fluorescens. Надо отметить, что данные филогенетического анализа соответствуют таксономической классификации этих вирусов. Так, бактериофаги P. aeruginosa внутри первого кластера группируются отдельно и относятся к роду Phikmvvirus, в том числе и исследуемый бактериофаг vB_PaeA-551w. Исключением является бактериофаг Pseudomonas phage LKA1, который на филогенетическом древе представлен отдельной веткой. Данный бактериофаг по таксономической классификации относится к другому роду Stubburvirus. Что касается бактериофагов Бактериофаги vB_PaeA-55-1w и vB_PaeM-198 обозначены красным цветом Pseudomonas agarici, Pseudomonas putida и Pseudomonas fluorescens в составе первого большого кластера, то каждый из них тоже занимает индивидуальную ветвь на филогенетическом древе и относится к отдельному таксону, а именно родам Kirikabuvirus, Tunggulvirus и Kantovirus соответственно. Второй большой кластер на филогенетическом древе сформирован бактериофагами, хозяева которых — бактерии видов Pseudomonas syringae, Pseudomonas tolaasii, Pseudomonas putida, Pseudomonas fluorescens, а также бактерии рода Pseudomonas, чья видовая принадлежность не установлена. Во втором кластере филогенетические подгруппы также соответствуют родам бактериофагов, входящих в них, а именно Pifdecavirus, Ghunavirus, Troedvirus, Pollyceevirus, Phutvirus, Napahaivirus, Pijolavirus, Pifdecavirus, Bifseptvirus, Uliginvirus. Следует отметить, что несколько родов внутри семейства имели хозяев, принадлежащих к разным видам: Bifseptvirus — хозяева P. syringae и P. tolaasii; Ghunavirus — хозяева P. fluorescens, P. putida, P. syringae.
Бактериофаги P. aeruginosa семейства Myoviridae были представлены в различных участках филогенетического древа и соответствовали семи родам: Baikalvirus, Citexvirus, Elvirus, Nankokuvirus, Pakpunavirus, Pbunavirus и Phikzvirus (см. рис. 1Б).
Исследуемый бактериофаг vB_PaeM-198 располагается на филогенетическом древе совместно с другими 26 бактериофагами рода Pbunavirus. Бактериофаги P. fluorescens, P. putida, P. syringae Myoviridae семейства относятся к 6 различным родам: Chakrabartyvirus, Flaumdravirus, Noxifervirus, Otagovirus, Plaisancevirus и Tabernariusvirus.
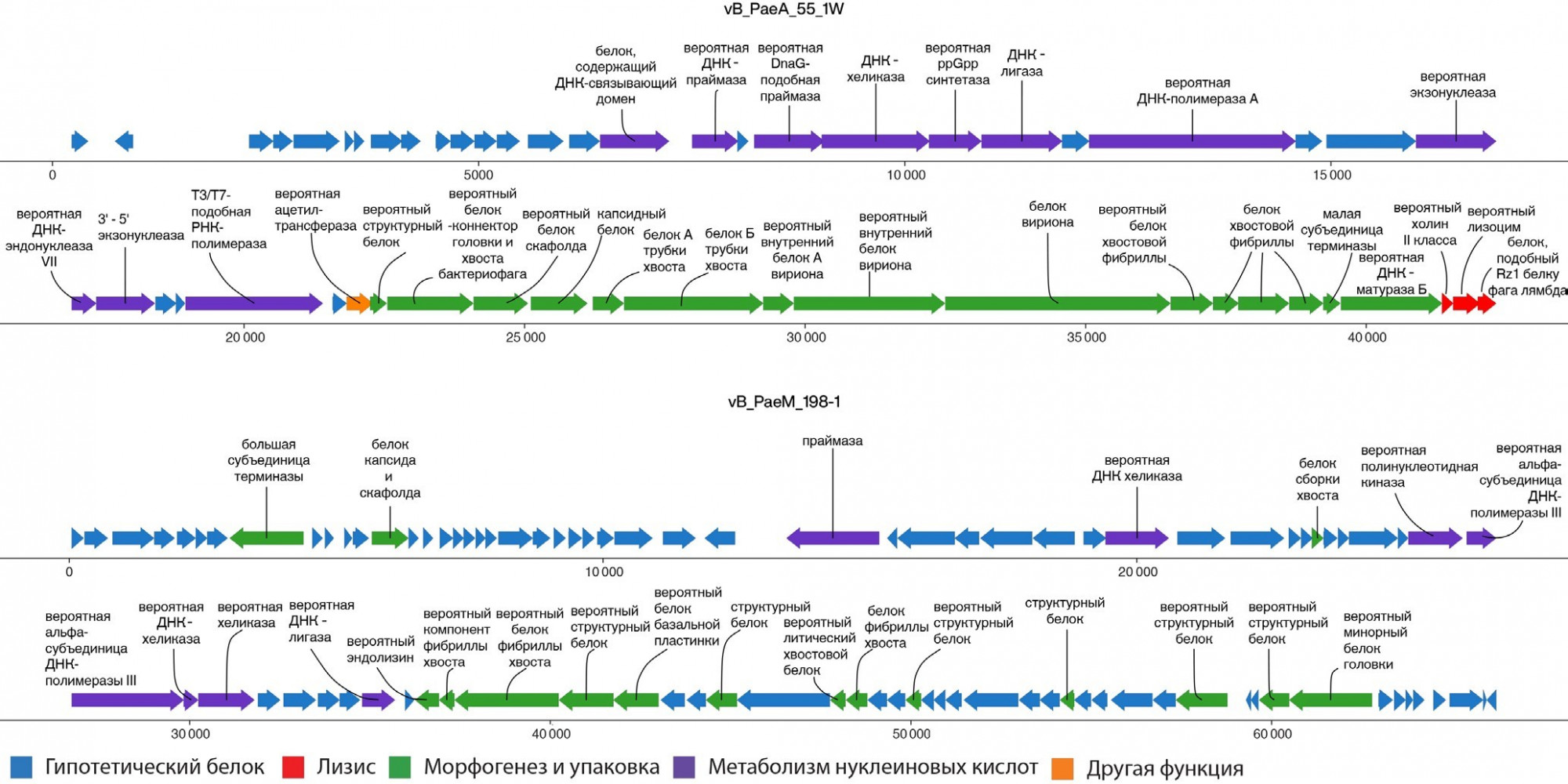
Модульное строение бактериофагов vB_PaeA-55-1w и vB_PaeM-198
Для описания геномной организации бактериофагов vB_PaeA-55-1w и vB_PaeM-198 был проведен анализ функциональных модулей геномов. Надо отметить, что число генов, функции которых удалось установить, было больше для бактериофага vB_PaeA-55-1w (n = 30/52, 58%), чем для vB_PaeM-198 (n = 24/95, 25%) (рис. 2). В ходе анализа для обоих исследуемых фагов была установлена локализация модулей метаболизма нуклеиновых кислот и морфогенеза и упаковки. Расположение модуля лизиса было установлено для бактериофага vB_PaeA-55-1w. Прицельный поиск генов известных бактериальных токсинов и различных интеграз в составе геномов vB_ PaeA-55-1w и vB_PaeM-198 не выявил их наличия.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
На фоне кризиса антибиотиков, связанного с распространением бактерий с МЛУ и ЭЛУ, бактериофаги P. aeruginosa начинают все чаще применять в терапевтической практике. Для терапии инфекций, вызванных P. aeruginosa, наиболее часто используют представителей семейств Autographiviridae и Myoviridae [22, 23]. В рамках данной работы из природных источников были выделены бактериофаги семейств Autographiviridae (vB_PaeA-55-1w) и Myoviridae (vB_PaeM198), обладающие широким спектром действия (47,5% и 50% соответственно), сопоставимым с бактериофагами P. aeruginosa соответствующих семейств [24, 25]. Следует отметить, что бактериофаги вызывали лизис различных штаммов, что может увеличить эффективность терапии при использовании фагового коктейля, включающего в себя оба исследуемых бактериофага.
Следует также подчеркнуть, что бактериофаги вызывали лизис штаммов, относящихся к различным сиквенс-типам, в том числе и штаммов ST235 (n = 1), ST244 (n = 4) и ST395 (n = 1). Изоляты, принадлежащие к данным сиквенс-типам, относятся к наиболее распространенным по всему миру, часто связаны со вспышками инфекционных заболеваний, а также характеризуются повышенным уровнем устойчивости к антибактериальным препаратам [26]. Интересным представляется дальнейшее исследование литической активности бактериофагов vB_PaeA-55-1w и vB_PaeM-198 на коллекции штаммов S. aureus, относящихся к сиквенстипам высокого эпидемического риска.
По современным требованиям, предъявляемым к терапевтическим препаратам, необходимо их детальное описание, а в случае бактериофагов — обязательно проводить определение их геномных последовательностей [27], что связано с необходимостью подтвердить вирулентный характер бактериофагов, а именно отсутствие генов интеграз в геноме. Умеренные бактериофаги для терапии не применяют по причине того, что они могут способствовать переносу генов бактериальных токсинов, а также детерминант устойчивости к антибиотикам в популяции бактерий [27]. Кроме того, с целью оценки безопасности применения бактериофага в терапии осуществляют поиск генов известных токсинов в составе генома [27].
Для исследуемых бактериофагов обоих семейств было продемонстрировано типичное модульное строение генома [24, 28], включающее модуль метаболизма нуклеиновых кислот, модуль морфогенеза и упаковки. Кроме того, для vB_PaeA-55-1w (семейство Autographiviridae) была установлена локализация модуля лизиса. В случае бактериофага vB_PaeM-198 (семейство Myoviridae) генов, обладающих высокой степенью идентичности с известными генами лизинов или холинов, обнаружено не было. Нужно отметить, что для обоих исследуемых бактериофагов модуля лизогении, включающего гены интеграз, обнаружено не было, что подтверждает их вирулентный характер. Также не было выявлено известных генов токсинов, что делает исследуемые бактериофаги потенциально пригодными для терапии.
На основании данных филогенетического анализа бактериофагов Pseudomonas spp, относящихся к семействам Autographiviridae и Myoviridae, было показано, что бактериофаги P. aeruginosa вне зависимости от семейства формируют отдельные кластеры на филогенетических деревьях, соответствующие родам Phikmvvirus и Stubburvirus (семейство Autographiviridae), а также Baikalvirus, Citexvirus, Elvirus, Nankokuvirus, Pakpunavirus, Pbunavirus, Phikzvirus, Phitrevirus (семейство Myoviridae). Данный факт указывает на то, что бактериофаги P. aeruginosa видоспецифичны. Отдельно надо отметить, что бактериофаги, описанные ранее и применяемые в фаготерапии (phiKMV, PPA-ABTNL, MPK6, RLP), в том числе ΦNH-4 (Pbunaviruses) и PAK_P1 (Pakpunavirus), показали свою эффективность на животных моделях [29, 30] и также кластеризуются совместно с исследуемыми бактериофагами vB_PaeA-55-1w и vB_PaeM-198.
ВЫВОДЫ
Исходя из проведенного анализа, бактериофаги vB_ PaeA-55-1w и vB_PaeM-198 можно рекомендовать для потенциального применения в фаготерапии, в том числе и инфекций, вызванных штаммами P. aeruginosa с МЛУ.

- Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnetet DL, et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis. 2018; 18 (3): 318–27.
- Horcajada JP, Montero M, Oliver A, Sorlí L, Luque S, GómezZorrilla S, et al. Epidemiology and treatment of multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa infections. Clin Microbiol Rev. 2019; 32 (4).
- Kuzmenkov AY, Trushin IV, Vinogradova AG, Avramenko AA, Sukhorukova MV, Malhotra-Kumar S, et al. AMRmap: an interactive web platform for analysis of antimicrobial resistance surveillance data in Russia. Front Microbiol. 2021; 12: 620002.
- Pena C, Cabot G, Gómez-Zorrilla S, Zamorano L, OcampoSosa A, Murillas J, et al. Influence of virulence genotype and resistance profile in the mortality of Pseudomonas aeruginosa bloodstream infections. Clin Infect Dis. 2015; 60 (4): 539–48.
- 2020 Antibacterial agents in clinical and preclinical development: an overview and analysis. Geneva: World Health Organization, 2021. Available from: https://www.who.int/publications/i/ item/9789240021303.
- Gordillo Altamirano FL, Barr JJ. Phage therapy in the postantibiotic era. Clinical Microbiology Reviews. 2019; 32 (2): e00066-18. DOI: 10.1128/CMR.00066-18.
- Акимкин В. Г., Дарбеева О. С., Колков В.Ф. Бактериофаги: исторические и современные аспекты их применения: опыт и перспективы. Клиническая практика. 2010; 1 (4): 48–54.
- Chen F, Cheng X, Li J, Yuan X, Huang X, Lian M, et al. Novel lytic phages protect cells and mice against Pseudomonas aeruginosa infection. J Virol. 2021; 95 (8): e01832-20. DOI: 10.1128/ JVI.01832-20.
- Jault P, Cheng X, Li J, Yuan X, Huang X, Lian M, et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): a randomised, controlled, double-blind phase 1/2 trial. Lancet Infect Dis. 2019; 19 (1): 35–45.
- Oechslin F. Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses. 2018; 10 (7): 351. DOI: 10.3390/v10070351.
- Купцов Н. С. и др. Эффективность препаратов бактериофагов против патогенов группы ESKAPE. Вестник РГМУ. 2020; (3): 19–26.
- Mazzocco A, et al. Enumeration of bacteriophages using the small drop plaque assay system. Methods Mol Biol. 2009; 501: 81–85.
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Pr. 1989, 2200 p.
- Seemann T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics. Oxford University Press. 2014; 30 (14): 2068–9.
- Aziz RK, et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genomics. BioMed Central. 2008; 9: 75.
- Laslett D. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004; 32 (1): 11–16.
- Lefkowitz EJ, et al. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018: 46 (1): 708–17.
- Meier-Kolthoff JP, Göker M. VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics. 2017; 33 (21): 3396–404.
- Lefort V, Desper R, Gascuel O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol Biol. 2015; 32 (10): 2798–800.
- FigTree. Available from: http://tree.bio.ed.ac.uk/software/figtree/. (Дата обращения: 16.07.2021).
- Enright MC, et al. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol. 2000; 38 (3): 1008–15.
- Alvi IA, et al. RLP, a bacteriophage of the family Podoviridae, rescues mice from bacteremia caused by multi-drug-resistant Pseudomonas aeruginosa. Arch Virol. 2020. 165 (6): 1289–97.
- Farlow J, et al. Complete Genome Sequences of 10 Phages Lytic against Multidrug-Resistant Pseudomonas aeruginosa. Microbiol Resour. 2020. 9: 29.
- Alvi IA, Asif M, Rehman S. A single dose of a virulent bacteriophage vB PaeP-SaPL, rescues bacteremic mice infected with multi drug resistant Pseudomonas aeruginosa. Virus Res. 2021; 292: 198250.
- Adnan M, et al. Isolation and characterization of bacteriophage to control multidrug-resistant Pseudomonas aeruginosa planktonic cells and biofilm. Biologicals. 2020; 63: 89–96.
- Treepong P, et al. Global emergence of the widespread Pseudomonas aeruginosa ST235 clone. Clin Microbiol Infect. 2018; 24 (3): 258–66.
- Principi N, Silvestri E, Esposito S. Advantages and Limitations of Bacteriophages for the Treatment of Bacterial Infections. Front Pharmacol. 2019; 10: 513.
- Guo Y, Chen P, Lin Z, Wang T. Characterization of Two Pseudomonas aeruginosa Viruses vB_PaeM_SCUT-S1 and vB_PaeM_SCUT-S2. Viruses. 2019; 11 (4): 318. DOI: 10.3390/ v11040318.
- Alemayehu D, Casey PG, McAuliffe O, Guinane CM, Martin JG, Shanahan F, et al. Bacteriophages φMR299-2 and φNH-4 can eliminate Pseudomonas aeruginosa in the murine lung and on cystic fibrosis lung airway cells. MBio. 2012; 3 (2): e00029-12. DOI: 10.1128/mBio.00029-12.
- Debarbieux L, Leduc D, Maura D, Morello E, Criscuolo A, Grossi O, et al. Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J Infect Dis. 2010; 201 (7): 1096–104.